Study of the crystal structure of organic compounds and pharmaceutical substances, polymorphism, phase transitions, and supramolecular organization.
1. Study of Polymorphism in Crystals of Biologically Active Substances.
Polymorphism in the crystals of biologically active compounds is one of the key research areas of the department. Specifically, this topic was the focus of work funded by a NFDU grant. It is known that different polymorphic modifications of solid forms of pharmaceutical substances can exhibit fundamentally different activities, which ultimately depend on the crystalline organization of the molecules. Therefore, monitoring and predicting polymorphism in both current and potential pharmaceutical substances is extremely important for the development and production processes of pharmaceutical drugs.
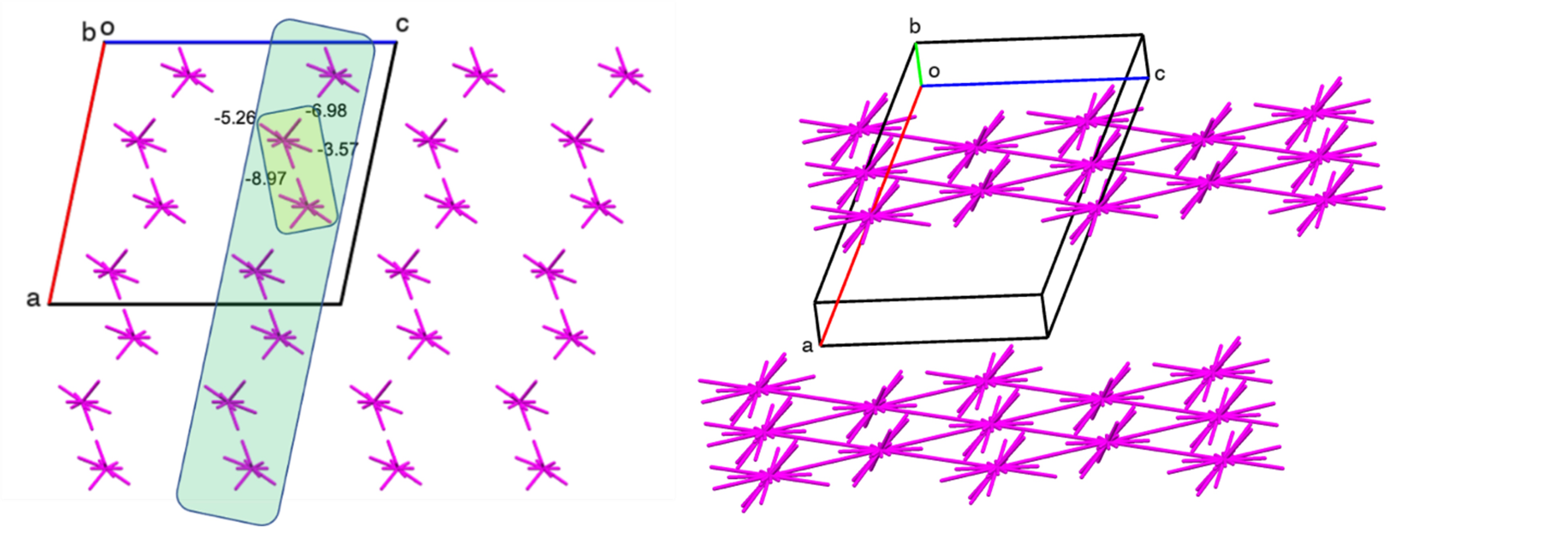
One striking example is the well-known drug piracetam, which has five polymorphic modifications. Despite the drug's popularity, including in research, the reasons for the transitions between forms and their relative stability were not clear. The study of the crystal structures of polymorphic modifications using energy vector diagrams, developed and implemented within the department, revealed both similarities and significant differences between the II and V modifications of piracetam. The transition between these forms is induced by the application of pressure (Fig. 1):
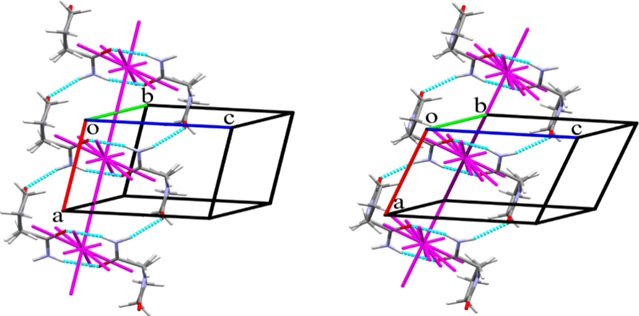
Both structures have a similar energetic configuration, yet form II exhibits greater energetic anisotropy (layered structure), while in form V, the energy of intermolecular interactions is distributed more evenly (a structure closer to columnar). The transition between these structures can be triggered by displacement along certain crystallographic directions without disrupting the crystal structure; the energetic parameters of this displacement were evaluated using quantum-chemical computational methods (Figure 2).
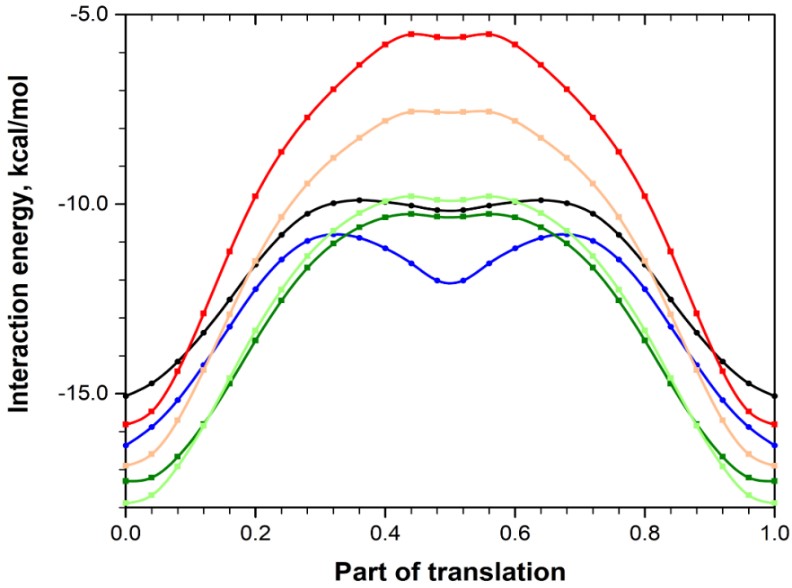
The presence of a secondary minimum in the energy profile curve of the polymorphs indicates an easy shift of the dimeric building unit of both crystals along the base layer, which is a prerequisite for the polymorphic transition between the forms. The energy barriers for the shift turned out to be small and quite accessible at low pressure values – for example, those achieved during the mechanical processing of substances in pharmaceutical manufacturing.
Another example of a pharmaceutical compound with different known polymorphic modifications is ibuprofen: it has one stable crystal form, while a second (unstable) polymorphic modification and an unstable amorphous solid form of the substance are also known. To study this case, an analysis of the conformational space of the individual molecule was applied by scanning torsion angles, which revealed significant potential for free rotation of certain groups, a prerequisite for conformational changes and, consequently, crystal organization under pressure. At the same time, known experimental studies showed that torsion angles differ only slightly across the various polymorphic modifications. Modeling the shear deformation showed no local minima on the energy profile, similar to those in piracetam, and significantly higher shear energies, which provides the basis for explaining experimental data regarding the number of polymorphic modifications of the substances and the possibility of transitions between them under pressure.
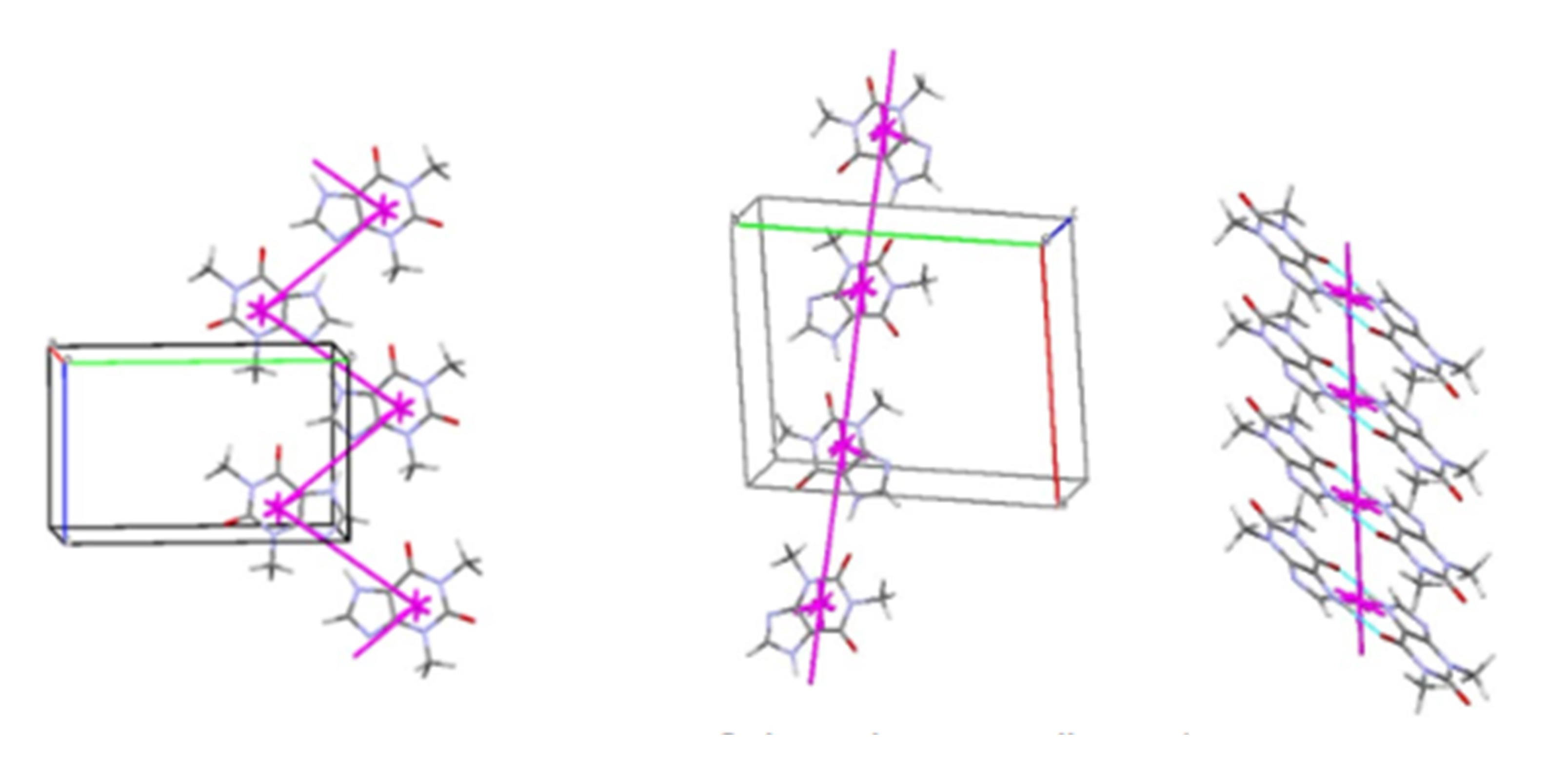
An example of a compound whose biological activity may change during pharmaceutical manipulation is the analgesic drug 1-Allyl-4-hydroxy-2,2-dioxo-N-(4-methoxyphenyl)-1H-2λ6,1-benzothiazine-3-carboxamide – a pyrroxicam analogue with a similar molecular structure. The structure of two polymorphic modifications of the compound and key intermolecular interactions were analyzed: the results of the energy vector diagram method revealed a fundamental difference in the structure (Figure 3).
The study using powder X-ray diffraction analysis revealed that both polymorphs are stable under normal conditions for a long period. However, a transition from the layered structure to the columnar structure is observed during grinding in the cuvette (Figure 4).
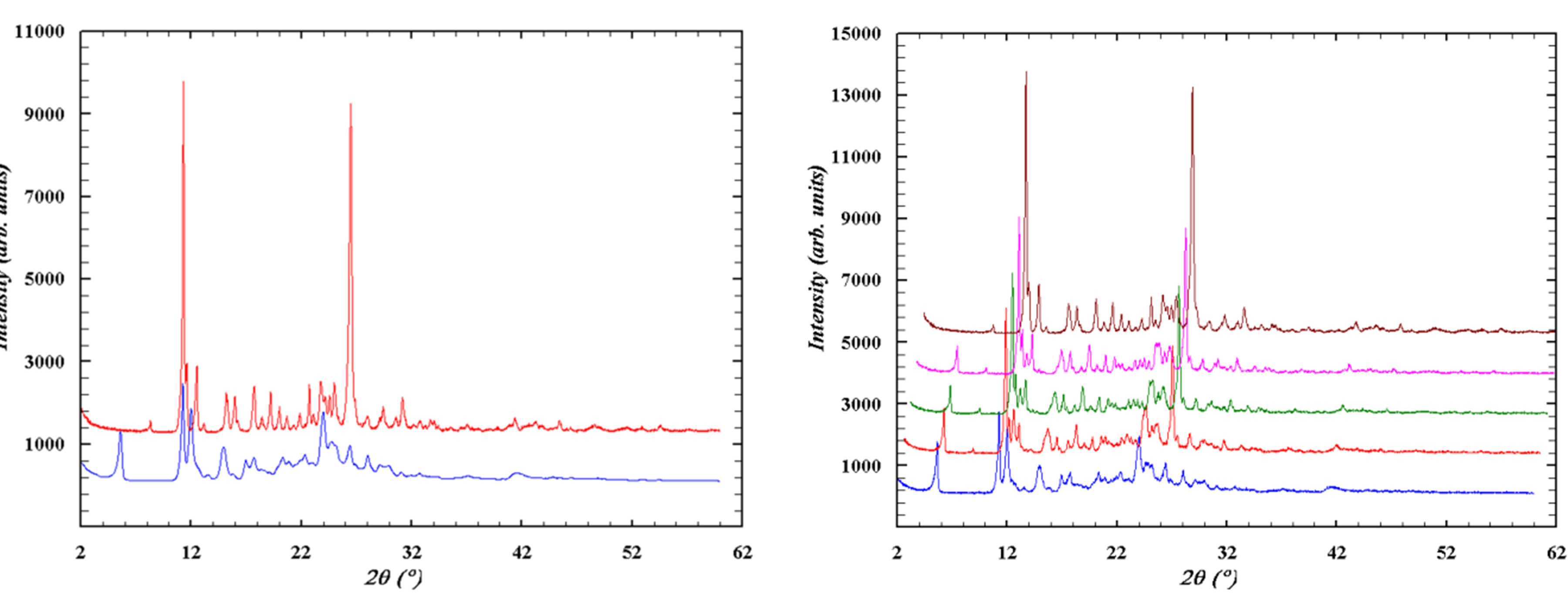
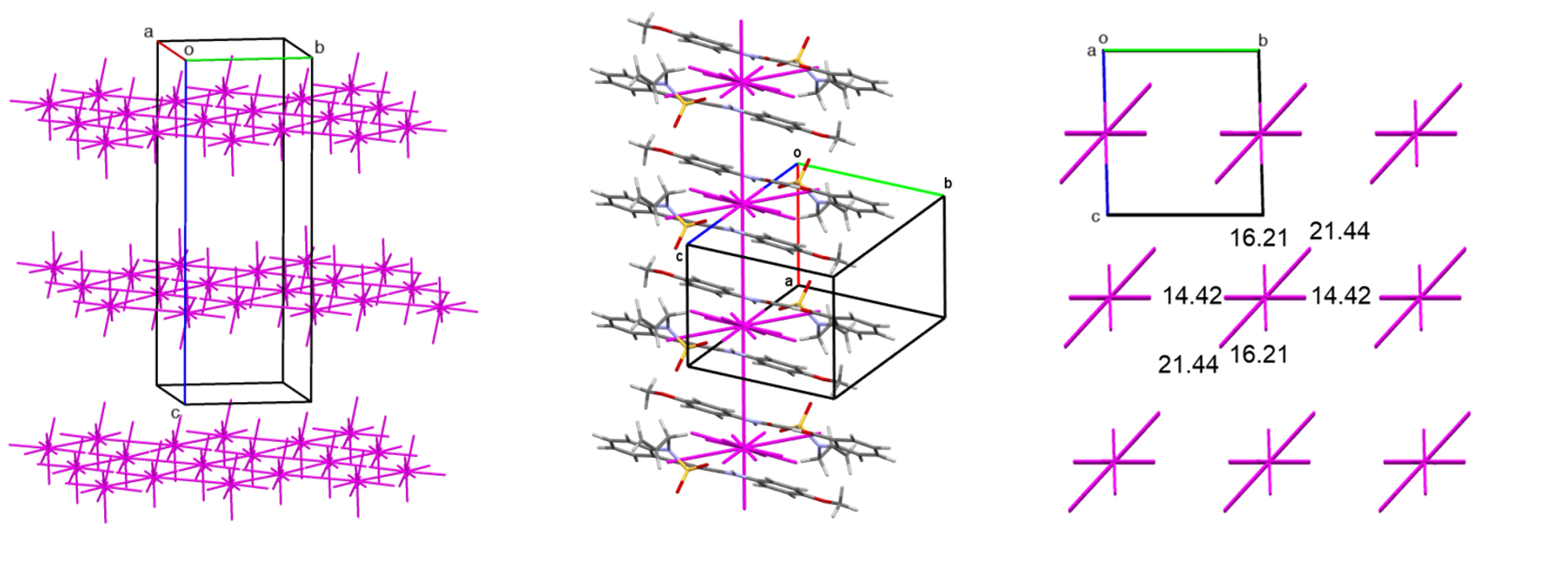
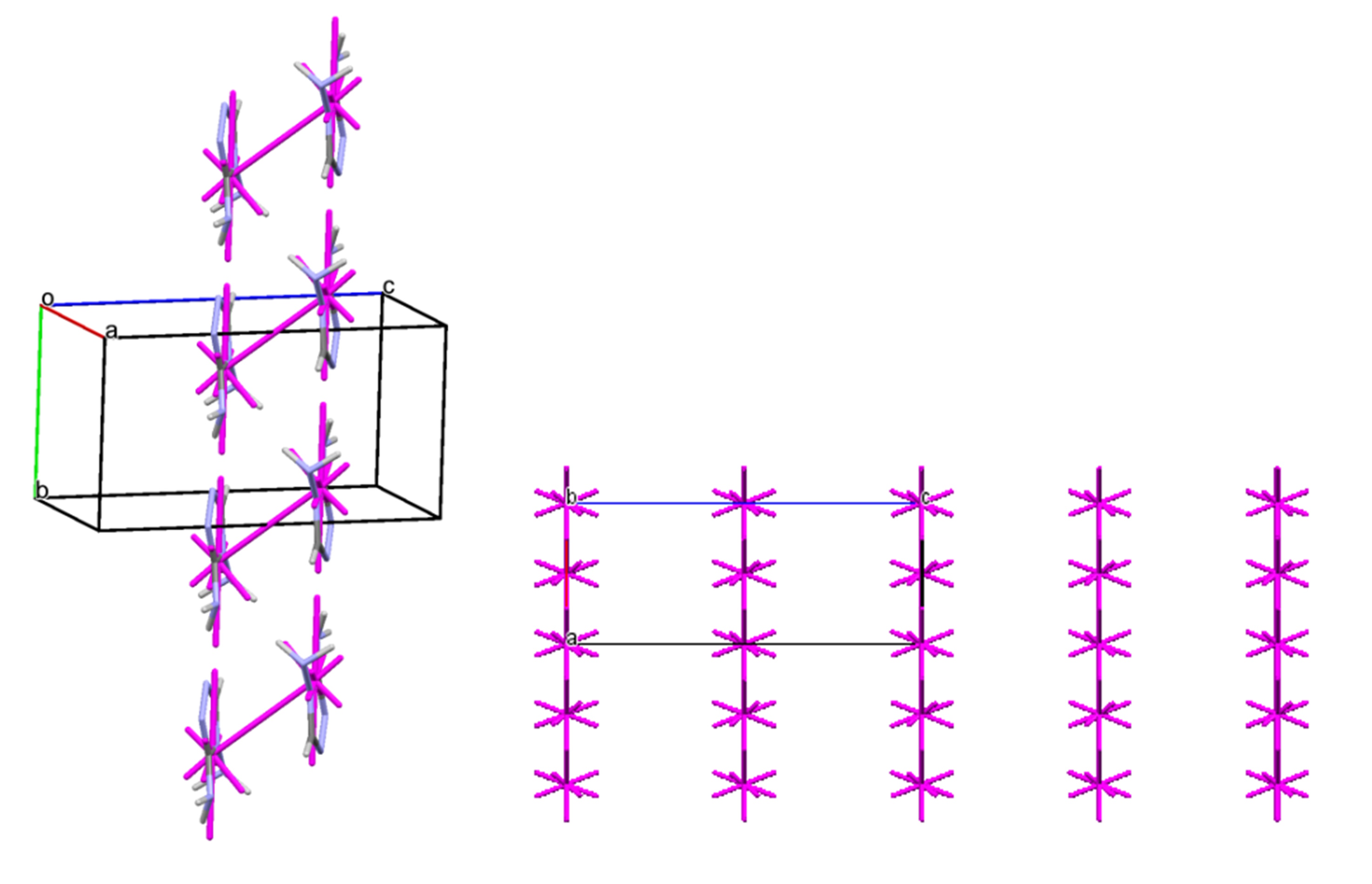
A similar situation is observed during mechanical impact on a potential biologically active substance from the oxazole series (3-(1-(tert-butoxycarbonyl)azetidin-3-yl)-1,2-oxazole-4-carboxylic acid). Comprehensive studies of the compound using single-crystal and powder X-ray diffraction methods, as well as quantum-chemical analysis of intermolecular interactions, allowed for the identification and description of the polymorphic transition between structures during grinding—mechanical loading similar to the crushing process during tablet production in the pharmaceutical industry (Figures 5, 6).
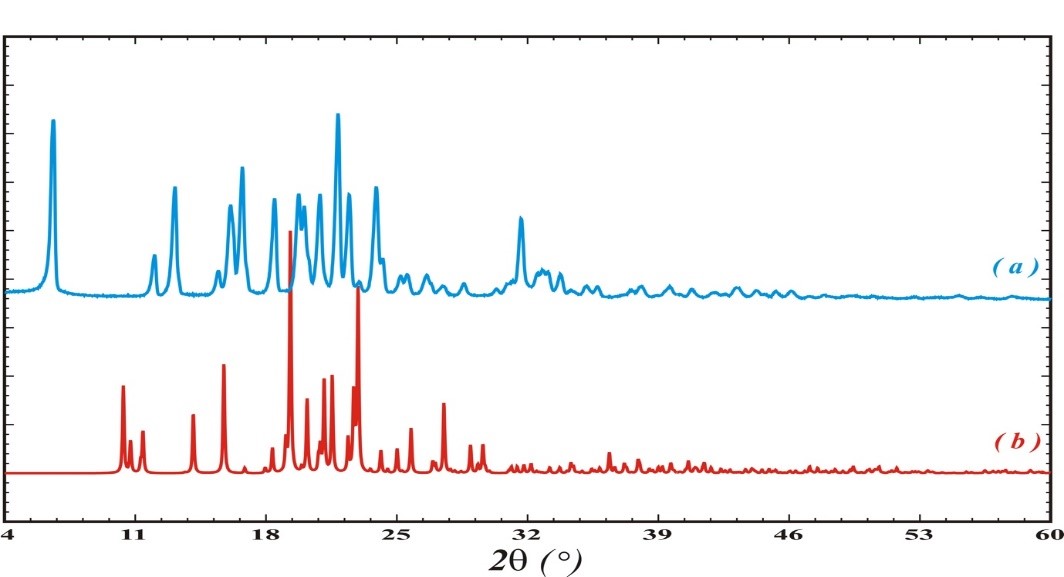
Structure b has a columnar structure (Figure 6, left), while structure a, which forms after grinding, is based on a layered packing of centrosymmetric dimers as the primary structural motif (Figure 6, right).
he biologically active compound theophylline also demonstrates a variety of polymorphic modifications. The crystal structures of three known modifications, as well as the monohydrate and a co-crystal with iodine, have been studied. All polymorphic modifications are variations of columnar-layered structures (energy vector diagrams are shown in Figure 7), with significant differences primarily in the degree of folding of these columns and their mutual arrangement.
Diaminotriazoles, which serve as a key structural unit for many biologically active molecules, also exhibit diverse modes of crystal packing. Despite having limited conformational space, they possess numerous functional groups capable of forming intermolecular interactions of various types. Studies of two polymorphic modifications of diaminotriazoles revealed different crystal structures from the perspective of energy vector diagrams (Figure 8, monoclinic modification – left, and orthorhombic – right). The difference between the polymorphs is attributed to weak interactions involving amino groups. Thermal expansion studies showed minor differences between the polymorphic modifications, and based on lattice energy calculations, the monoclinic modification is predicted to be more stable.
2. Analysis of the Crystal Structures of Organometallic Complexes
Organometallic complexes are of great significance for the development of biological, physical, and pharmaceutical chemistry, and they can often only be obtained and studied in crystalline form. They exhibit a wide variety of intermolecular interactions, resulting in diverse molecular packing arrangements. Studies in the department focused on structures with ligands derived from the biologically active class of carbamoylphosphates. These compounds are analogs of β-diketones but exhibit different coordination behavior.
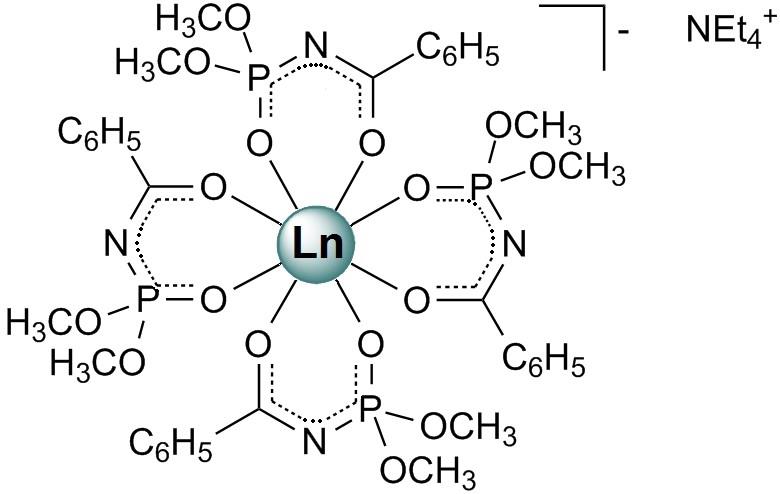
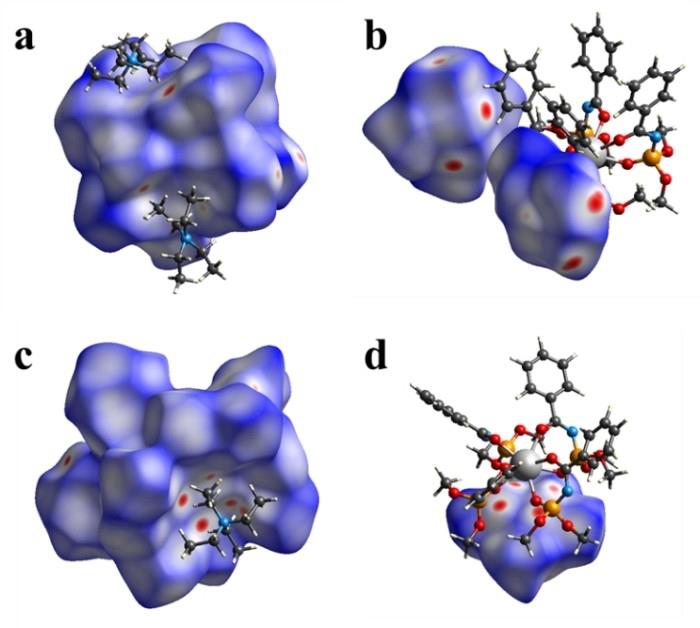
Structural characteristics of new dimethyl-N-benzoylamidophosphates (HL) based on tetrakis complexes of lanthanides NEt4[LnL4] (Ln³⁺ = Nd, Gd, Tb) were examined (Figure 9). Although their coordination polyhedra are similar, their structures demonstrate different crystal packing modes. Analysis of intermolecular interactions using the Hirshfeld surface approach revealed the dominant role of H...H contacts, while the contributions of O...H/H...O and C...H/H...C were significantly smaller (Figure 10). A detailed analysis of intermolecular interactions highlighted the dominant role of non-local interactions, such as stacking and nonspecific interactions, in forming the crystal packing.
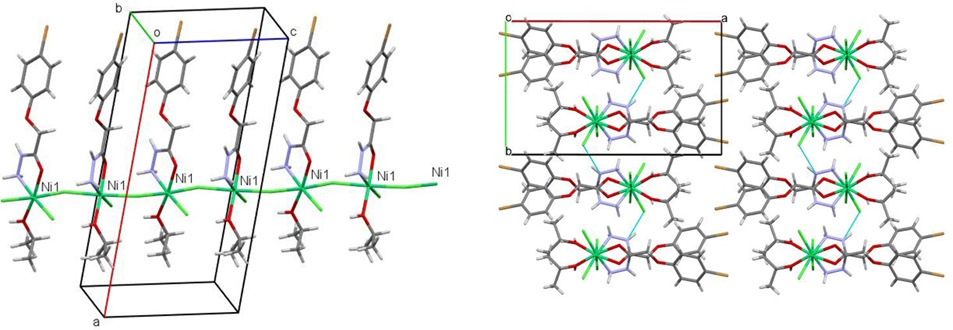
Another research focus involved hydrazide derivatives, which are also potential pharmaceutical agents. The structure of 2-(4-bromophenoxy)acetohydrazide and its complex with nickel(II) chloride was studied. In the crystalline phase, the compound forms polymeric chains through coordination bonds, which are assembled into layers via weaker intermolecular interactions (Figure 11).
Additionally, the structure and stereochemical factors governing the formation of five cation-anion complexes with similar Fe(II), Co(II), Ni(II), Cu(II) 1,10-phenanthroline cations and various complementary malato-Ge/Sn anions [M(phen)3]2[{Sn(HMal)2(Mal)}Cl] (M = Fe (2), Co (3), Ni (4), Cu (5)) and [Co(phen)3]2[{Sn(HMal)(Mal)2] were investigated (Figure 12).
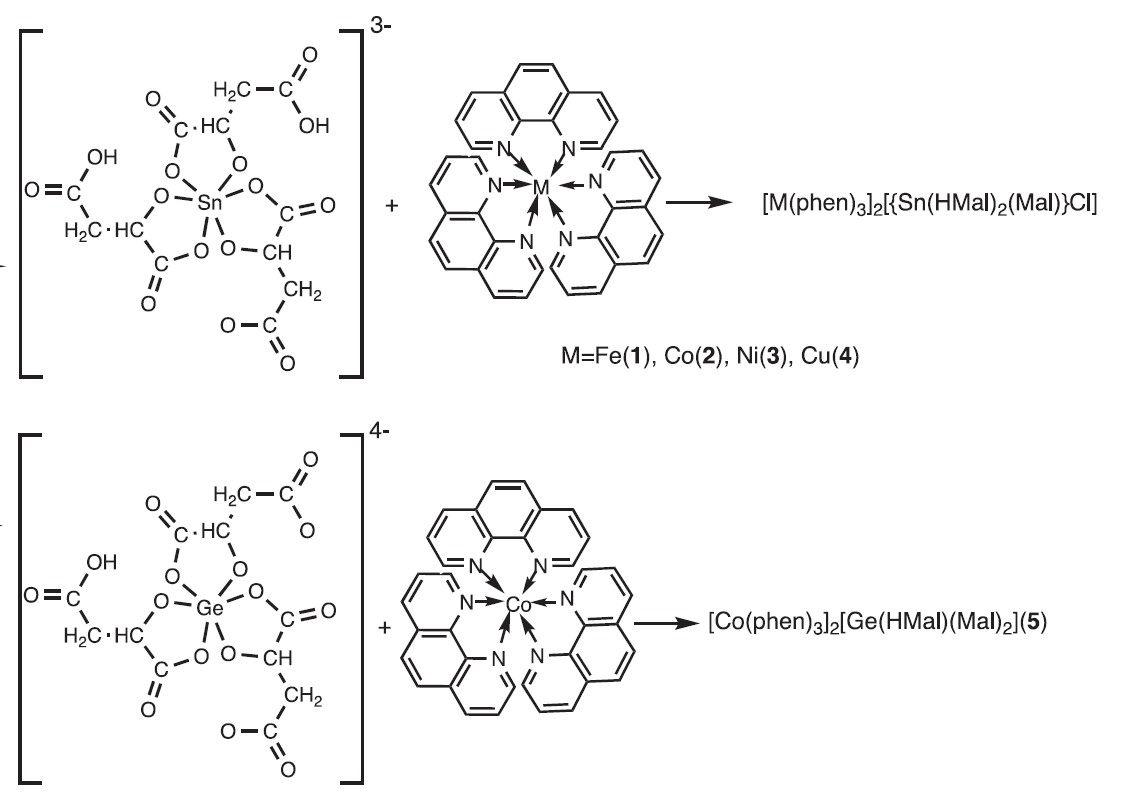
Analysis of intermolecular interactions confirmed the key contribution of H...O/O...H, H...H, H...C/C...H, and O...C/C...O interactions to the formation of the Hirshfeld surface for all anions, while additional C...C interactions played a more significant role in cations, confirming the presence of π-π interactions in their structures.
3. Crystal Structures of Aromatic and Non-Aromatic Nitrogen-Containing Compounds and Their Response to Mechanical Pressure
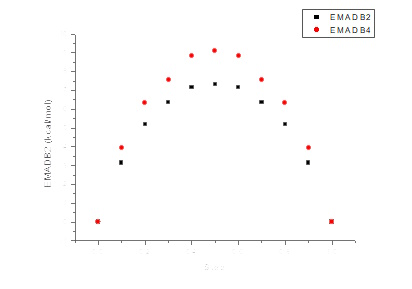
Quantum-chemical studies were conducted on a series of nitrogen-containing compounds, including nitrobenzene derivatives and conjugated non-aromatic nitrogen-containing compounds exhibiting explosive properties. For example, simulations of structural changes in the isomers 2-amino-1,3-dinitrobenzene (MADB-2) and 4-amino-1,3-dinitrobenzene (MADB-4), which are structurally similar to sym-triaminotrinitrobenzene and have layered crystal structures, revealed that isotropic mechanical pressure up to 20 GPa reduces unit cell dimensions by 5–17%.
MADB-2 and MADB-4 exhibit pronounced anisotropic responses, with compression occurring predominantly along the direction of the primary structural motif—stacked columns (similar to TATB behavior). In MADB-2, this corresponds to the (100) direction, while in MADB-4, the stacked columns are oriented along (10-1), with parameters a and c primarily absorbing the additional pressure.
The basic structural motifs in the crystals of MADB-2 and MADB-4 evolve progressively under pressure: the columnar structure becomes more pronounced, as stacking interactions lose less energy than other types of interactions. However, no fundamental structural transition occurs, similar to the behavior of TATB.
Shear energy profiles demonstrate moderate deformation energy barriers along the path of least resistance between adjacent layers (Figure 13).
Another area of study involved the polymorphic modifications of trinitrotoluene (TNT) to identify potential transitions between them under pressure. The basic structural motifs in the orthorhombic and monoclinic crystal modifications are similar, differing mainly in secondary structural organization, while both are based on stacked columns. Studies under isotropic pressure revealed that excess compression energy is primarily absorbed by layers approaching each other.
However, unlike triaminotrinitrobenzenes and monoaminodinitrobenzenes, which exhibit a similar trend, in the case of TNT, the interaction between these layers is not associated with stacking and results from a combination of numerous local and non-local intermolecular pair interactions. At the same time, unlike planar nitroamino compounds, TNT exhibits significantly lower amplitude of unit cell parameter changes and anisotropy of these changes, which indicates the absence of dominant interactions with significant buffering properties, such as stacking.
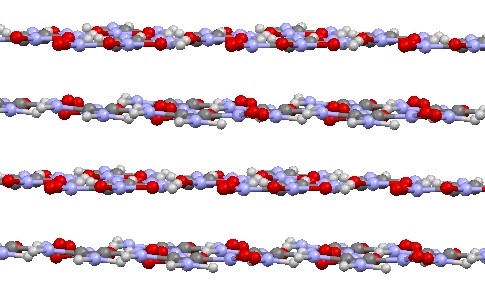
An interesting aspect is the comparison of the behavior of aromatic nitrobenzenes and non-aromatic 1,1-diamino-2,2-dinitroethylene (FOX7). FOX7 has several known polymorphic modifications, the most stable of which have a layered structure. The monoclinic and orthorhombic modifications exhibit a wavy-layered structure, which is very similar to the structures of the isomeric diaminotrinitrobenzenes discussed earlier (Figures 14a-b). In contrast, the triclinic modification has a planar-layered structure, which resembles that of sym-triaminotrinitrobenzene (Figure 15).
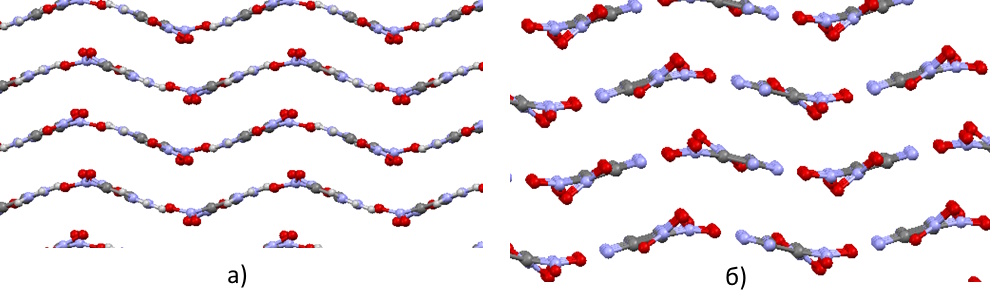
Increasing pressure leads to slightly different effects on wavy-layered and planar-layered structures, as well as reveals certain differences in the behavior of aromatic nitrobenzenes and non-aromatic diamino-dinitroethylene. Although the primary mechanism for compensating mechanical pressure in all studied structures remains compression in the direction perpendicular to the layer planes, wavy-layered structures (both aromatic and non-aromatic) exhibit a noticeable tendency to increase the degree of layer folding. In non-aromatic diamino-dinitroethylene, this tendency is somewhat more pronounced (the wavelength decreases by 14-16%, while for aromatic structures, the reduction reaches only 9-13%).
Planar structures tend to maintain planar layers. However, in the non-aromatic structure of triclinic diamino-dinitroethylene, a significantly higher degree of molecular deformation under pressure is observed (the maximum angles of nitro group deviation from the molecular plane reach 36° at 20 GPa, while the TATB molecule retains its planar structure with maximum nitro group deviation angles of up to 15°).
4. Investigation of Phase Transitions and Hydration/Dehydration Processes of Inorganic Compounds Using Powder X-Ray Diffraction
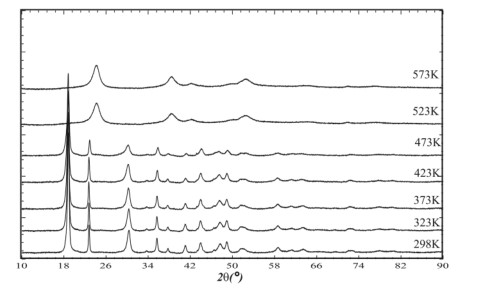
The thermal decomposition of nickel oxalate dihydrate ultimately results in a disordered crystalline form of anhydrous nickel carbonate. Powder diffraction patterns recorded during the decomposition process allowed for the construction of dehydration diagrams and determination of the main steps of the solid-state transformation (Figures 16–17).
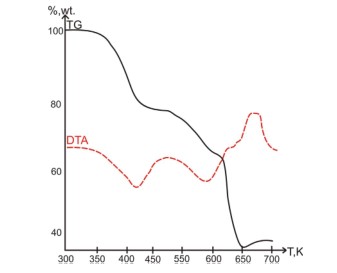
5. Pharmaceutical Studies
In collaboration with pharmaceutical companies in Ukraine, X-ray diffraction measurements of commercial drug samples are routinely performed to determine the phase composition of substances.